Authors
Hanyan Li, Mike Barber, Jingrang Lu, Ramesh Goel
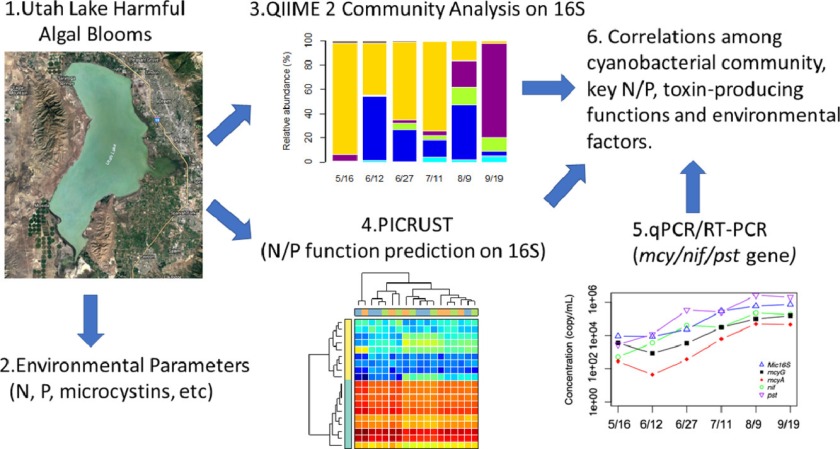
The current study reports the community succession of different toxin and non-toxin producing cyanobacteria at different stages of cyanobacterial harmful algal blooms (CyanoHABs) and their connectivity with nitrogen and phosphorus cycles in a freshwater lake using an ecogenomics framework. Comprehensive high throughput DNA sequencing, water quality parameter measurements, and functional gene expressions over temporal and spatial scales were employed. Among the cyanobacterial community, the lake was initially dominated by Cyanobium during the months of May, June, and early July, and later primarily by Aphanizomenon and Dolichospermum depicting functional redundancy. Finally, Planktothrix appeared in late August and then the dominance switched to Planktothrix in September. Microcystis aeruginosa and Microcystis panniformis; two species responsible for cyanotoxin production, were also present in August and September, but in significantly smaller relative abundance. MC-LR (0.06–1.32 µg/L) and MC-RR (0.01–0.26 µg/L) were two major types of cyanotoxins detected. The presence of MC-LR and MC-RR were significantly correlated with the Microcystis-related genes (16SMic/mcyA/mcyG) and their expressions (r = 0.33 to 0.8, p < 0.05). The metabolic analyses further linked the presence of different cyanobacterial groups with distinct functions. The nitrogen metabolisms detected a relatively higher abundance of nitrite/nitrate reductase in early summer, indicating significant denitrification activity and the activation of N-fixation in the blooms dominated by Aphanizomenon/Dolichospermum (community richness) during nutrient-limited conditions. The phosphorus and carbohydrate metabolisms detected a trend to initiate a nutrient starvation alert and store nutrients from early summer, while utilizing the stored polyphosphate and carbohydrate (PPX and F6PPK) during the extreme ortho-P scarcity period, mostly in August or September. Specifically, the abundance of Aphanizomenon and Dolichospermum was positively correlated with the nitrogen-fixing nif gene and (p < 0.001) and the PPX enzyme for the stored polyphosphate utilization (r = 0.77, p < 0.001). Interestingly, the lake experienced a longer N-fixing period (2–3 months) before non-fixing cyanobacteria (Planktothrix) dominated the entire lake in late summer. The Provo Bay site, which is known to be nutrient-rich historically, had early episodes of filamentous cyanobacteria blooms compared to the rest of the lake.