Authors
Jin Zhou, Yong-min Lao, Jun-ting Song, Hui Jin, Jian-ming Zhu, Zhong-hua Cai
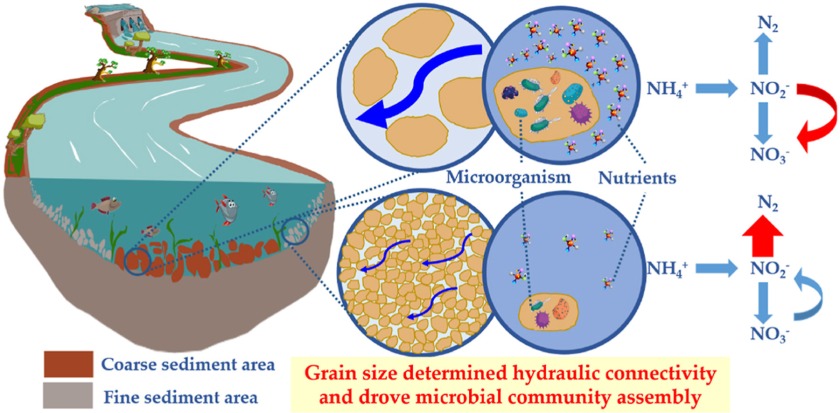
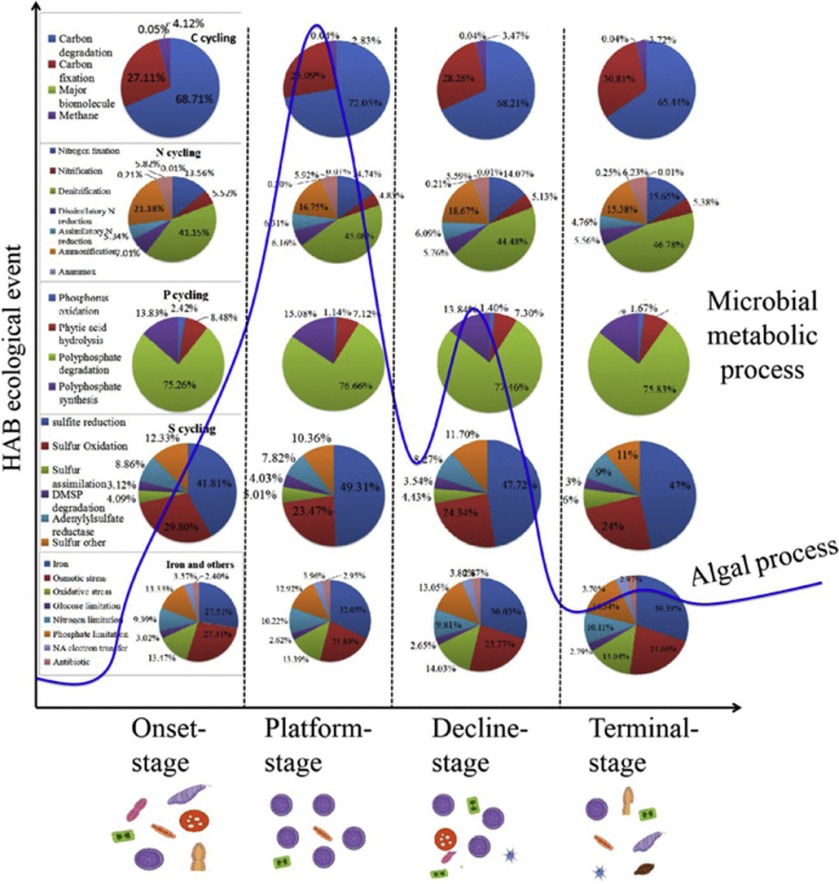
Elucidating the interactions between algae and associated microbial communities is critical for understanding the mechanisms that mediate the dynamic of harmful algal blooms (HABs) in marine environment. However, the microbial functional profiles and their biogeochemical potential in HABs process remains elusive, especially during a complete natural HAB cycle. Here, we used pyrosequencing and functional gene array (GeoChip) to investigate microbial community dynamics and metabolic potential during a natural dinoflagellate (Noctiluca scintillans) bloom. The results shown that bacterioplankton exhibited significant temporal heterogeneity over the course of the bloom stages. Microbial succession was co-driven by environmental parameters and biotic interactions. The functional analysis revealed significant variations in microbial metabolism during matter cycling. At bloom onset-stage, metabolic potential associated with iron oxidation and transport was elevated. Carbon fixation and degradation, denitrification, phosphorus acquisition, and sulfur transfer/oxidation were significantly enhanced at the plateau stage. During the decline and terminal stages, oxidative stress, lysis of compounds, and toxin degradation & protease synthesis increased. This work reveal phycosphere microorganisms can enhanced organic C decomposition capacity, altered N assimilation rate and S/P turnover efficiency, and balancing of the Fe budget during HAB process. The ecological linkage analysis has further shown that microbial composition and functional potential were significantly linked to algal blooms occurrence. It suggest that structural variability and functional plasticity of microbial communities influence HAB trajectory.